Vanessa Camelier* e M ª Gabriela C. Cavalcante*
IDENTIFICAÇÃO: A.S.F., 43 anos, sexo feminino, casada,
servente, natural de Pompéia (SP) e procedente de Conceição
do Jacuípe (BA).
H.M.A: Paciente refere que, a cerca de 7 anos, cursou com discreta protrusão
do globo ocular esquerdo, associada a sensação de “areia
no olho”, dor, caracterizada como em compressão, e prurido
ocasional. Relata ainda dores nos MMSS e MMII, astenia, alopecia, palpitações,
tremores nas extremidades, instabilidade emocional e intolerância
ao calor. Paciente negou alterações do sono ou ponderais,
dispnéia e disfagia. No primeiro momento, procurou oftalmologista
para avaliação (I.D.: Tireoidopatia a esclarecer), o qual
solicitou uma consulta com endocrinologista (nov/97). Foram realizados
ultrassonografia, cintilografia (vide Exames Complementares) e função
tireoidiana e prescrito Propanolol 40 mg via oral (1/2 comprimido de 12
em 12 hrs, sendo depois aumentado para 1/2 comprimido 3X/dia). O quadro
era de oftalmopatia de Graves, mas não estava aflorado. Inclusive
a captação ainda não estava elevada.
I.S: Nega rouquidão e precordialgia; refere prurido vaginal e dispareunia.
ANTECEDENTES PESSOAIS
- Médicos: Paciente alérgica a carne de porco, poeira,
amendoim, camarão, trigo, lã, fungos; em tratamento.
- Ginecológicos: Laqueadura tubária há 14 anos.
- Familiares: Tias paterna e materna com DM, irmã cardiopata
+ HAS, nega tireoidopatia e histórico de amaurose na família.
Não soube informar sobre glaucoma.
HÁBITOS DE VIDA: Nega etilismo, ex-tabagista há 3 anos
(05 cigarros/dia). Passado de moradia em casa de taipa.
EXAME FÍSICO
Geral : Paciente em BEG, LOTE, eupnéica, afebril, mucosas coradas,
escleróticas anictéricas. Peso = 59 Kg / Altura = 1,54
/ IMC = 24,9. PA = 120/80mmHg PR = 88bpm.
Pescoço: tireóide aumentada e de superfície regular.
AR: MVBD sem RA. ACV: BRNF em 2T, sem sopros.
Abdome: RHA +, discretamente doloroso à palpação
em hipogástrico e FIE.
Extremidades: sem edemas e acianóticas, com tremores finos.
Reflexos: patelar e aquileu presentes e simétricos.
Gânglios: pré-auriculares e sub-mandibulares impalpáveis.
Exame Oftalmológico: Presença de exoftalmia à esquerda.
Pálpebras, cílios e conjuntivas sem alterações.
Pupila fotorreagente, câmara anterior ampla, MOE sem alterações.
Oftalmoscopia mostrou estruturas normais.
EXAMES COMPLEMENTARES
USG DE TIREÓIDE : tireóide discretamente aumentada de
volume, ecotextura heterogênea com pequeno nódulo sólido,
hipoecogênico, medindo 6 mm de diâmetro localizado no LD.
LD = 5,3 X 2,0 X 1,6 cm LE = 5,0 X 1,5 X 1,6 cm (Out/97);
CINTILOGRAFIA Captação de I 131 = 20% em 24 hrs ( VR:
8 à 35% ). Conclusão : normocaptação tardia
do radioiodo pela tireóide. Impressão Diagnóstica
: Bócio Difuso . (Nov/97)
EXAMES LABORATORIAIS
| SETEMBRO / 97 |
T4 = 12,8 ug/100ml (VN = 4,5 – 13,0)
T3 = 2,0 ng/100ml (VN = 0,5 – 2,1)
TSH = 0,03 uUI/ml (VN = 0,10 – 9,0) (dim.)
AntiTPO = 8,86 U/ml (VN< 0,3 U/ml) (aum.) |
| NOVEMBRO / 97 |
TRH/TSH
coleta basal 0,58(0,8 – 5,0); 30 min após 0,8 mUI/ml;
60 min após 0,47 mUI/ml |
SUSPEITAS DIAGNÓSTICAS: Doença Auto-imune; Oftalmopatia
de Graves.
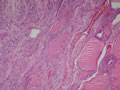
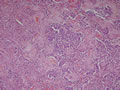
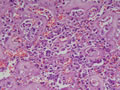
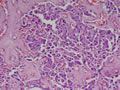
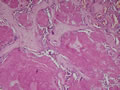
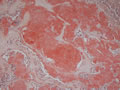
BIÓPSIA:
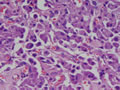
| Figura 1: Pequeno aumento. |
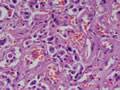
| Figura 2: Pequeno aumento. |
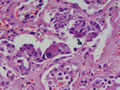
| Figura 3: Médio aumento. Idem. |
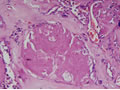
| Figura 4: Médio aumento. |
| Figura 5: Grande aumento. Idem. |
| Figura 6: Grande aumento. Idem. |
| Figura 7: Grande aumento. Idem. |
| Figura 8: Pequeno aumento. Idem. |
| Figura 9: Pequeno aumento. Idem. |
| Figura 10: Pequeno aumento. Idem. |
| Figura 11: Pequeno aumento. Idem. |
EVOLUÇÃO
26/03/98: Paciente com suspeitas diagnósticas
de Doença de Graves relata que com a medicação
o tamanho dos olhos está diminuindo e alguns dos sintomas tiveram
melhoras. Terapêutica: Propanolol 40 mg (1/2 comprimido VO de
08/08 hrs), um remédio para alergia e vacinas para alergia. PA
= 110/95mmHg PR = 100bpm. Conduta: Manter propanolol e, quando a paciente
trouxer a captação e o mapeamento, programar iodoterapia.
07/05/98: Relata dor em MMII, indisposição,
nervosismo, palpitações, sensação de calor.
Refere ainda dor em região cervical esquerda com irradiação
para HTE, nervosismo, indisposição, sudorese, palpitações.
Informa ainda que acha que os olhos estão de igual tamanho, pois
o menor aumentou. Terapêutica: Propanolol 40 mg (1/2 comprimido
VO de 08/08 hrs) e Polaramine 02 mg 3X/dia. PA = 90/70mmHg PR = 128bpm.
Conduta: Orientou-se a realização de novos exames de controle
hormonal, nova cintilografia e captação. Após os
resultados, encaminhamento para dose terapêutica de Iodo.
01/07/99: Paciente permaneceu 14 meses sem acompanhamento
médico, além de ter suspendido a medicação
prescrita (propanolol) por conta própria há 12 meses e
não ter feito uso da dose treapêutica de Iodo radioativo.
Relata que cursou com piora dos sintomas, referindo muita astenia, sudorese
profusa, tremores nas extremidades, nervosismo, mas com sono preservado.
Referiu também regressão da exoftalmia. Ao exame físico,
constatou-se a pele quente; tireóide aumentada de tamanho com
nódulo raso de +/- 1 cm em LD; tremores finais de extremidades,
com calor irradiante; e reflexos patelares exaltados. PA = 120/90mmHg
PR = 88bpm. Conduta: Solicitou-se, então, novos exames para avaliação
da evolução do hipertireoidismo (hormônios, USG,
cintilografia).
01/10/99: Foram solicitadas novas dosagens de T3,
T4 L, TSH e nova cintilografia da tireóide com duas captações
em 24 hrs. Tapazol 20 mg foi mantido.
04/05/00: Paciente em uso de Tapazol 20 mg, refere
manutenção de nervosismo, sudorese. Diz ter melhorado
da insônia. Ao exame físico, tireóide aumentada
de 2X, fibroresistente, superfície irregular, sem nódulos
ou gânglios cervicais palpáveis. Extremidades sem edemas
ou tremores. Pele um pouco “pegajosa”. PA = 110/70mmHg PR
= 88bpm. Realizada nova cintilografia da tireóide (11/04/00)
com resultado a considerar. Conduta: Foi prescrito dose terapêutica
de I 131 .
16/11/00: Paciente segue em uso de Tapazol 20 mg.
Não fez dose terapêutica de I 131 (contra-indicada pela
medicina nuclear). Evolui relatando nervosismo discreto, palpitações
e tremores. Às vezes sudorese moderada. Nega insônia, ritmo
intestinal 01 vez/dia, mas refere ganho ponderal neste último
período (cerca de 4 Kg). Ao exame físico, tireóide
aumentada de 2X, fibroresistente, sem nódulos palpáveis
ou gânglios cervicais aumentados. Extremidades sem edemas e com
tremores finos. Pele quente, irradiando calor. Reflexos pouco exacerbados.
Olhos sem proptose e MOE sem comprometimento. PA=110/70mmHg PR = 88bpm.
Realizada nova cintilografia da tireóide (18/08/00). T4 L (18/08/00)
= 1,57 (VN = 0,8 – 2,0). Conduta: Diminuição da
dose do Tapazol para 10 mg/dia. Prescrito complexo B. Solicitação
de nova função tireoidiana e dosagem de magnésio.
| |
1998 |
1999 |
2000 |
| T4 Livre |
2,7 ug/100ml
(VN=0,8–2,0)
|
2,55
(VN=0,68–1,76)
|
0,76
(VN=0,68– 1,76) |
| TSH |
1,63 uUI/ml
(VN = 0,4 – 4,5)
|
0,19
(VN=0,34 - 5,6)
|
0,01
(VN=0,34–5,6) |
| T3 |
*** |
1,69
(VN = 0,8 – 2,0)
|
0,65
(VN=0,8–2,0) |
| AAM |
Positivo (1:1600) |
Positivo (1:6400)
|
Positivo (1:6400) |
| AntiTPO |
*** |
*** |
*** |
01/10/99: realizada cintilografia da tireóide
com Tc99 que mostrou glândula tópica, morfologia preservada,
contornos bem definidos, aumentada de grau leve, homogênea, sem
lesões nodulares ao estudo. Conclusão: Bócio difuso
com captação 24 hrs do Iodo 131 nos limites superiores
à normalidade. Captação: 44%/24 hrs.
11/04/00: mostrou glândula tópica, de
dimensões aumentadas, com radiofármaco alta intensidade,
heterogênea com predomínio do LD, sem evidências
de lesões nodulares. Conclusão – discreto bócio
difuso hiperfuncionante. Captação 41% em 2 hrs e 88 %
em 24 hrs, mostrando hipercaptação precoce e tardia.
18/08/00: cintilografia com Tc99 mostrou tireóide
hiperfuncionante com dimensões nos limites superiores à
normalidade. Captação: 50 % em 24 hrs (hipercaptação
tardia).
05/04/01: Paciente em uso de Tapazol 10mg/dia e Propanolol
40mg/dia, retorna negando insônia, nervosismo, tremores, sudorese,
alteração do ritmo intestinal. Refere ganho ponderal no
período. Ao exame físico, tireóide aumentada de
2X, fibroresistente, sem sopro ou frêmito, sem lindonodos palpáveis.
Abdome doloroso à palpação de abdome inferior,
sem visceromegalia ou dor à descompressão. Pele e reflexos
normais. Conduta: Foi orientada a suspensão das medicações
e solicitadas novas dosagens hormonais com 45 dias e ultrassonografia.
21/06/01: Paciente sem uso de quaisquer medicações,
retorna relatando melhora do quadro de nervosismo, tremor, insônia
e sudorese. Relata ainda intolerância ao frio no período
e astenia. Nega sonolência. Ao exame físico, tireóide
pouco aumentada de tamanho, endurecida, sem nódulos palpáveis.
Pele ressecada e extremidades sem edemas. PA = sem tensiômetro
disponível PR = 84 bpm. Mg = 2,1 (VN = 1,9 – 2,5). Conduta:
Aguarda-se resultados da função tireoidiana e USG da tireóide.
19/07/01: Paciente cursa assintomática, sem
uso de medicações. Vem trazer resultados de exames. Conduta:
acompanhamento a cada 6 meses.
06/03/03: Paciente sem uso de medicações,
apenas refere utilização esporádica de Diazepam
5 mg. Retorna referindo episódios de insônia, aumento dos
ciclos menstruais para 5 dias e ritmo intestinal diário. PA =
90/60mmHg PR = 80bpm. Trouxe resultados de exames. Ultrassonografia
da tireóide (12/03/03) mostra LD (1,7 X 4,3 X 1,3 cm ) com textura
heterogênea às custas de imagem hipoecóica em 1/3
médio com 1,5 cm. LE (1,1 X 4,0 X 1,0 cm ) com textura homogênea
e contornos regulares. Conclusão: nódulo sólido
em LD. Conduta: Solicitou-se PAAF e aguarda-se a função
tireoidiana. Hemograma e bioquímica normais.
29/05/03: Paciente com diagnósticos de Doença
de Graves (há +/- 5 anos, tratada com medicação)
e Nódulo Tireoidiano em LD. Paciente retorna sem novas queixas
e fazendo uso esporádico de Diazepam 5 mg e outra medicação
que não soube referir. Ao exame físico, ausência
de adenomegalia cervical. PA = 100/80mmHg PR = 72bpm. Conduta: Solicitou-se
novamente a PAAF.
28/08/03: Paciente sem uso de medicações,
retorna negando alteração do apetite e sonolência.
Ritmo intestinal 3/3 dias. Referiu artralgia em quirodáctilos.
Ao exame físico, tireóide de superfície irregular
às custas de nódulo em LD, fibro-resistente e móvel.
PA = 110/60mmHg PR = 80bpm. Conduta: aguarda-se PAAF.
20/11/03: Ao exame físico, PA = 110/70mmHg
PR = 76bpm. Resultado da PAAF (20/08/03) diagnosticou nódulo
proliferativo epitelial em lobo direito com indicação
de conduta cirúrgica. Conduta: Paciente foi encaminhada para
a cirurgia.
12/05/04: Paciente evolui hemodinamicamente estável,
tendo sido submetida à Tireoidectomia Subtotal secundária
a Nódulo Tireóideo. Recebe alta em boas condições
e orientada, sendo solicitado o seu retorno, ao hospital, para acompanhamento.
| |
2001 |
2002 |
2003 |
| T4 Livre |
10,2
(VN=12–22 pmol/l) |
1,5 ng/dl
(VN =0, 75 – 1,8 ) |
1,4
(VN=0,8–1,9 ng/dl) |
| TSH |
5,77
(VN=0,34– 5,6) |
1,74
(VN = 0,35 – 5,3) |
2,2
(VN=0,4–4,0 uUI/ml) |
| T3 |
1,11
(VN= 0,8 – 2,0) |
*** |
93
(VN=70–170 ng/dl) |
| AAM |
Positivo (1:600) |
*** |
*** |
| AntiTPO |
*** |
*** |
216 UI/ml (VR<15) |
RESUMO E COMENTÁRIOS: Trata-se de uma paciente do sexo feminino,
43 anos de idade, com história de exoftalmia unilateral, em globo
ocular esquerdo, associada a sintomas de tireotoxicose. Após consulta
com oftalmologista, foi encaminhada ao ambulatório de endocrinologia
onde foi diagnosticada Doença de Graves. A ultrassonografia mostrou
tireóide aumentada discretamente de tamanho
com pequeno nódulo sólido, hipoecogênico, medindo
6 mm de diâmetro em lobo direito. A cintilografia mostrou normocaptação
tardia do radioiodo pela tireóide.
Paciente evoluiu com manutenção dos sintomas durante seis
anos. Em Fev/03, foi realizada nova ultrassonografia que mostrou nódulo
sólido em LD com 1,5 cm. PAAF realizada em Set/03 mostrou nódulo
proliferativo em lobo direito. Paciente foi encaminhada para cirurgia,
tendo realizado Tireoidectomia Subtotal secundária a Nódulo
Tireóideo. O exame anátomo-patológico revelou Carcinoma
Medular da Tireóide em lobo direito (1,5 cm) e Tireoidite Linfocítica.
Aspectos Gerais
Embora seja a malignidade endócrina mais comum, o Câncer
de Tireóide é uma doença rara, correspondendo a cerca
de 1% de todos os casos de neoplasia maligna. [1]
Este tipo de câncer é representado por um grupo heterogêneo
de tumores, que possuem uma grande variabilidade em seu comportamento
biológico, aparência histológica e resposta à
terapia. [2]
Os tumores malignos da Tireóide são provenientes de dois
grupos celulares de origens embriológicas distintas. As células
C, neuroendócrinas, produtoras de calcitonina, cujo tumor é
o Carcinoma Medular, e as células Foliculares produtoras de T4
e tireoglobulina que originam os tumores bem diferenciados e os indiferenciados.
[2]
O Carcinoma Medular da Tireóide ( CMT ) representa 3 a 10% dos
tumores malignos da glândula. Ele consiste de neoplasia de células
parafoliculares ou células C, produtoras de calcitonina. [1]
O CMT é mais freqüente na forma esporádica ou não
– hereditária (75-80%) [3] . Nos demais casos, é uma
doença hereditária autossômica dominante com alto
grau de penetrância e variabilidade de expressão podendo
fazer parte de 3 síndromes clínicas distintas dependendo
dos órgãos envolvidos : Neoplasia Endócrina Múltipla
(NEM), 2A e 2B, e Carcinoma Medular da Tireóide Familiar (CMTF)
[1] .
Patogênese
Estudos genético – moleculares realizados nos últimos
anos têm demonstrado o envolvimento do proto – oncogene RET
(rearranged during transfection) na maioria da formas hereditárias
de CMT e, em menor freqüência, em sua forma esporádica.
O principal fator predisponente é a radiação, particularmente
na infância. [1]
Mutações somáticas ( identificadas apenas no tumor
) no gene RET estão sendo implicadas na patogênese da forma
esporádica, mas estas mutações não parecem
ser uniformes entre as várias subpopulações de células
dentro de um mesmo tumor e mesmo de suas metástases. [1]. Para
alguns autores, esta heterogeneidade entre as células tumorais
de um mesmo paciente sugere que o CMT possa ter uma origem policlonal,
ou então que as mutações no proto – oncogene
RET não sejam eventos iniciais na tumorigênese do CMT esporádico,
mas sim de progressão. [1]
O gene RET codifica, no homem, um receptor transmembranoso da família
das tirosino-quinases ( TK, do inglês tyrosine-kinase ) para uma
família de ligantes, da qual o paradigma é o fator de crescimento
neurotrófico derivado de células da glia (GDNF, glial cell-derived
neurotropic growth factor).
Este gene está altamente expresso em alguns tecidos normais e
tumores derivados da crista neural. Ao ser induzido, o RET ativa múltiplas
e complexas vias de sinalização intracelulares ( RAS/MAP
quinase, fosfatidilinositol 3 – quinase ) que são responsáveis
pela ação do receptor RET no controle da proliferação,
sobrevida, diferenciação, interação e motilidade
celulares, ativação oncogênica e fenótipo neoplásico.
Entretanto os mecanismos moleculares pelos quais as mutações
levam à transformação neoplásica da célula
não estão bem definidos
Características do CMT Hereditário
Geralmente, o CMT é a primeira manifestação neoplásica
de famílias com NEM 2 tendo em vista sua elevada penetrância
e seu aparecimento precoce[5], principalmente no subtipo 2 A (75 %) [6]
. As associações da síndrome NEM 2 (Tabela 2) se
relacionam à origem embriológica única das células
C da Tireóide, da Medula Adrenal, da Paratireóide e do Plexo
Nervoso Autonômico Entérico, todos derivados da crista neural.
[3]
Entre os adultos afetados com MEN 2A, cerca de 90% deles apresentam
CMT, 50% feocromocitomas e 20-30% tumores da paratireóide [6].
A doença adrenomedular é usualmente multicêntrica
e bilateral, geralmente detectada após o aparecimento de CMT e
com taxa de malignidade inferior a 10% [3] . O hiperparatireoidismo ocorre
em, aproximadamente, 10 a 20% dos indivíduos com NEM 2A, acometendo
geralmente todas as glândulas paratireóides [3]. A lesão
histológica mais comumente observada nos estágios iniciais
da doença é a hiperplasia da glândula, porém,
se a doença é diagnosticada mais tardiamente, a lesão
adenomatosa se superpõe à hiperplasia [3] . Ressalta-se
que em casos de NEM 2A, a hipertensão pode ocorrer devido a presença
do feocromocitoma e a hipercalcemia e cálculos renais são
decorrentes do hiperparatireoidismo que pode estar presente.
A síndrome NEM 2B caracteriza-se por CMT (90%) de comportamento
muito agressivo, feocromocitoma (45%), ganglioneuromatose (100%) e hábitos
marfanóides (65%) [3] . Essa síndrome caracteriza-se por
um fenótipo único, que inclui ganglioneuromatose difusa
da língua, lábios, olhos e do trato gastrointestinal. As
fácies características são precocemente reconhecidas
durante a infância (neuromas da mucosa). O envolvimento gastrointestinal
pode causar diarréia e constipação intermitente,
dor abdominal, megacólon e, ocasionalmente, obstrução
intestinal. Outro aspecto fenotípico da NEM 2B é o hábito
marfanóide com dedos e extremidades longas, hiperextensão
de articulações e anormalidades epifisárias [3] .
O CMTF consiste na presença de CMT isolado em, pelo menos, quatro
membros da mesma família, e as outras formas de CMT hereditário
consistem no acometimento de dois ou três membros da mesma família
com CMT, sem a presença de feocromocitoma ou hiperparatireoidismo
[3] .
O CMT esporádico origina – se de uma única célula
C evoluindo para um tumor solitário [1] . A calcitonina, o principal
produto secretório das células C, é considerado o
marcador bioquímico para diagnóstico e seguimento pós
– operatório dos pacientes com CMT [1] . Outras ubstâncias
são produzidas, entre elas: CGRP, CEA, amilóide, somatostatina,
ACTH, prostaglandinas, catecolaminas, substância P dentre outras.
[1]
Clinicamente, se caracteriza como nódulo único ou massa
tireoidiana indolor, associada a linfadenopatia cervical ou a outros sintomas
locais. Geralmente, sem associações com outras endocrinopatias
múltiplas e o fenótipo do paciente é normal; a incidência
é maior na 4 ª e 6 ª décadas de vida. [2]
Alguns pacientes, entretanto, têm sintomas de doença invasiva
como dor, disfagia, rouquidão, sinais de comprometimento respiratório
e/ou sintomas de síndrome paraneoplásica devida à
secreção hormonal pelas células tumorais (rubor facial,
diarréia, dor óssea, síndrome de Cushing). [1]
Diagnóstico [1]
O diagnóstico é baseado na história clínica,
exame físico, calcitonina e CEA elevados, e biópsia por
agulha fina seguida de citologia. A investigação inicial
por imagem deve ser feita através da ultrassonografia cervical.
[1]
A confirmação do diagnóstico de CMT é realizada
pelo exame anátomo – patológico da peça cirúrgica,
que, macroscopicamente, apresenta-se como uma massa firme de tamanha variável,
podendo ser de difícil visualização até vários
centímetros. Localiza-se preferencialmente na área de maior
concentração de células C, ou seja, nos dois terços
superiores da tireóide, principalmente nas suas porções
mais laterais.
No CMT esporádico, o tumor é caracteristicamente unicêntrico.
Menos do que 20% dos pacientes com esta forma de CMT apresentam tumor
acometendo a glândula bilateralmente, e acredita-se que quando isto
ocorre é uma extensão do tumor primário. Já
nas síndromes hereditárias, o CMT é usualmente multicêntrico,
acometendo ambos os lobos tireoidianos em praticamente 100% dos casos.
À microscopia óptica, a aparência típica do
CMT é a de ninhos ou ilhas de células que podem ser fusiformes,
poligonais, arredondadas ou ovais; o citoplasma apresenta uma granulação
fina e os núcleos são uniformes e centrais, com uma relação
núcleo-citoplasmática baixa. O tumor comumente invade vasos
sangüíneos e linfáticos. O estroma tumoral caracteristicamente
contém amilóide, porém em 15-20% dos casos ele está
ausente. Este amilóide é formado a partir da calcitonina
ou, mais possivelmente, da pré-calcitonina
A dosagem de calcitonina não serve para a triagem diagnóstica,
já que existem outras doenças em que há elevação
de calcitonina, como o câncer de mama e pulmão e tireotoxicose,
o que deve ser levado em conta no diagnóstico diferencial. [5]
Como o CMT capta 131 I– MIBG (metaiodobenzilguanidina), a cintilografia
com esta substância pode ser usada não só para se
obter mais um dado diagnóstico, mas também na procura de
metástases em pesquisa de corpo inteiro (PCI). [1]
O diagnóstico de CMT pode ser dificultado não só
pela ausência de amilóide,
mas também pela presença de variantes histológicas
deste tipo tumoral, como variante papilífera, variante glandular/folicular,
variante oxifílica, variante de células gigantes, variante
de células pequenas, variante escamosa e outras.
A imunohistoquímica para calcitonina confirma o diagnóstico
de CMT. A marcação usualmente é intensa e atinge
quase todas as células tumorais, porém algumas lesões,
na maioria das vezes do subtipo de células pequenas, podem não
expressar imuno-reatividade para calcitonina.
Outro marcador sensível, mas não específico para
CMT, é o CEA, que é positivo em 88–100% dos casos.
Prognóstico
Geralmente, é mais agressivo que os carcinomas bem diferenciados
de tireóide. A principal via de disseminação é
linfática, sendo alta a freqüência de metástases
macro ou mesmo microscópicas para gânglios linfáticos
regionais. A incidência de metástases cervicais correlaciona-se
com o tamanho do tumor primário (9,5–30% nos de até
1cm / 55– 60% nos maiores de 2 cm). A virulência tumoral diminui
progressivamente nos casos de NEM 2B, CMT esporádico, NEM 2 A e
CMTF. [1]
Em fases mais avançadas da doença, outros sítios
comuns de metástases são mediastino, gânglios do hilo
pulmonar, pulmôes, fígado e ossos. [1]
A sobrevida em 10 anos é de cerca de 90% em doença confinada
à tireóide, 70% na presença de linfonodos comprometidos
e 20 % com metástases a distância.
Tratamento
O único tratamento eficaz do CMT, seja na forma esporádica
ou na hereditária, é a cirurgia. Nos casos onde não
existem metástases cervicais clinicamente detectáveis, considera
– se como obrigatória a tireoidectomia total associada ao
esvaziamento cervical central eletivo. Naqueles pacientes portadores de
metástases cervicais, o esvaziamento cervical deve ser total e,
se possível, funcional. [1]
A quimioterapia e a radioterapia apresentam resultados limitados no tratamento
do CMT. [3]
A radioimunoterapia é uma nova modalidade terapêutica,
na qual anticorpos monoclonais anti CEA são utilizados para o tratamento
do CMT, mas a remissão do quadro não foi atingida. [3]
A terapia gênica abre uma perspectiva promissora para o tratamento
do CMT. Distintas abordagens têm sido utilizadas: introdução
de genes supressores tumorais; transferência de genes que aumentam
a resposta imunológica contra o câncer (imunização
gênica) e terapias combinadas. [3]
Seguimento [1]
O seguimento dos pacientes pode ser realizado a partir das dosagens
séricas de calcitonina e antígeno carcinoembrionário,
que devem ser obtidas em torno de 2 meses após a tireoidectomia,
devido à meia-vida longa destes marcadores na circulação
sangüínea.
Os níveis normais de calcitonina são excelentes indicadores
de uma ressecção curativa, enquanto que níveis elevados
desse marcador indicam a necessidade do rastreamento de metástases.
BIBLIOGRAFIA:
- MAGALHÃES, P. K .R., et al.: Carcinoma Medular de Tireóide:
da Definição às Bases Moleculares. Arquivo
Brasileiro de Endocrinologia e Metabologia 47 (5): 515-525,
2003.
- FERRAZ, A. R., et al.: Diagnóstico e Tratamento do Câncer
da Tireóide. Projeto Diretrizes: 1-8, 2001.
- PUÑALES, M. K., et al.: Carcinoma Medular de Tireóide:
Aspectos Moleculares, Clínico – Oncológicos e Terapêuticos.
Arquivo Brasileiro de Endocrinologia e Metabologia
48 (1): 137-145, 2004.
- BERNE, R. M., LEVY, M. N. A Glândula Tireóide. In.:
Fisiologia, Rio de Janeiro: Guanabara Koogan S.A.,
3a ed., 1996, p.875-889.
- MACIEL, R. M. B.: Tumorigênese Molecular Tireoideana: Implicações
Para a Prática Médica. Arquivo Brasileiro de Endocrinologia
e Metabologia 46 (4): 381-391, 2002.
- PEIXOTO, M. C., et al.: Avaliação da Incidência
e do Comportamento do Câncer de Tireóide em Pacientes Com
Doença de Graves Submetidos a Tireoidectomia. Arquivo
Brasileiro de Endocrinologia e Metabologia 47 (5): 566-571,
2003.
- MURUSSI, M., et al.: Punção Aspirativa de Tireóide
com Agulha Fina em um Hospital Geral: Estudo de 754 Punções.
Arquivo Brasileiro de Endocrinologia e Metabologia
45 (6): 576-583, 2001.
- SILVA, T. F. G. M, POSSÍDIO, D. A. Distúrbios Funcionais
da Tireóide. In.: Condutas em Clínica
Médica, Rio de Janeiro: Editora Médica e Científica
Ltda., 2a ed., p.495-515, 2001.
- MACIEL, R. M. B.: O Laboratório no Diagnóstico e Seguimento
de Doenças Auto-imunes e Neoplásicas de Tireóide.
Arquivo Brasileiro de Endocrinologia e Metabologia
46 (1): 65-71, 2002.
- DEDIVITIS, R. A., et al.: Variante Oncocítica do Carcinoma
Medular. Relato de Caso. Arquivo Brasileiro de Endocrinologia
e Metabologia 48 (2): 315-317, 2004.
* Monitora e aluna do curso médico da Faculdade de Medicina da
Bahia, Universidade Federal da Bahia, cursando o 5º semestre. |