| página inicial > monitoria > feocromocitoma |
| MONITORIA | ||||||||||||||
| Discussão Anatomo-Clínica FEOCROMOCITOMA |
||||||||||||||
| Augusto Emanoel* e Jan Menezes Lopes* IDENTIFICAÇÃO:
T.S., 10 anos, sexo feminino, natural de Salvador e procedente de Salvador.
EXAMES COMPLEMENTARES
EXAMES LABORATORIAIS:
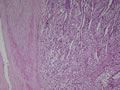
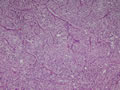
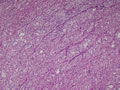
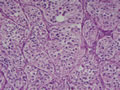
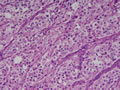
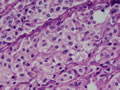
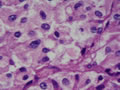
BIÓPSIA:
EVOLUÇÃO: No dia 06/05/04 a paciente foi submetida à cirurgia para retirada de massa tumoral retro-peritonial. Após a exérese da tumoração os níveis pressóricos da paciente apresentaram redução para o valor de 110x80mmHg no dia da alta(seis dias após a exérese), contudo mantendo picos hipertensivos durante o dia. O funcionamento intestinal foi normalizado. O diagnóstico anatomo-patológico foi de feocromocitoma em supra-renal esquerda. RESUMO E COMENTÁRIOS: Trata-se de uma paciente do sexo feminino,
10 anos de idade, com história de hipertensão arterial,
refratária a tratamento, taquicardia e vasoconstricção
periférica e edema de papila. Ultrassonografia abdominal revelou
tumoração retro-peritonial à esquerda cujo exame
anatomo-patológico revelou tratar-se de um feocromocitoma em supra-renal
esquerda. O quadro clínico observado no feocromocitoma é composto por cefaléia, sudorese e palpitação, náuseas, ansiedade, vaso constrição periférica, vômitos, dispnéia, dores abdominais, rubor facial e elevação da temperatura, associada à hipertensão refrataria a tratamento anti-hipertensivo, ocorrendo sintomas cardíacos (taquicardia, angina do peito, infarto agudo do miocárdio, miocardite, cardiomiopatia congestiva dilatada, arritmias), digestivo (obstipação), circulatórios (acidente vascular cerebral, hipotensão ortostática, choque), renais (proteinúria), pulmonares (edema agudo de pulmão por insuficiência cardíaca esquerda ou de etiologia não cardiogênica), Diabetes Mellitus (3). O diagnóstico laboratorial é obtido a partir do sumario de urina, dosando catecolaminas e seus metabólitos (ácido vanililmandélico, adrenalina, noradrenalina, metanefrinas, normetanefrinas), exame sanguíneo dosando adrenalina, noradrenalina, ácido vanililmandélico , níveis de hematócrito e VHS; sendo que todos esses achados estão elevados (4). No diagnóstico por imagem se utiliza: tomografia computadorizada, ultra-sonografia, ressonância magnética, positron emission tomography (PET) e mapeamento com 131I metaiodobenzilguanidina. O diagnóstico por testes funcionais é obtido a partir dos testes de supressão com clonidina e de estímulo com glucagon (3). O diagnóstico anatomo-patológico de malignidade só é estabelecido pela presença de metástase tendo ou não invasão da cápsula, a histologia com achados de anaplasia não é suficiente para determinação de malignidade nesta neoplasia (4). A taxa de sobrevida é de 5 anos após a cirurgia, em geral é maior que 95%, sendo a taxa de recorrência menor que 10%. Nos casos malignos a taxa de sobrevida de 5 anos não atinge 50%. A remoção completa do feocromocitoma produz cura da hipertensão em cerca de 75% dos pacientes. Nos demais casos, a hipertensão sofre recidiva; sendo geralmente bem controlada com uso de agentes anti-hipertensivos. Nesse grupo, a presença de hipertensão essencial subjacente ou de lesão vascular irreversível induzida por catecolaminas pode resultar em persistência da hipertensão. Em um seguimento de pacientes que realizaram cirurgia para retirada de feocromocitoma, 29% mantiveram-se hipertensos, sendo que 24% estavam em tratamento anti-hipertensivo (20% tinham níveis pressóricos controlados e os 4% restantes mantinham os níveis pressóricos sem controle) e 5% sem terapêutica. Aproximadamente 6% foram reoperados, por recorrência local ou evidência de metástase, numa média de 3,2 anos. Em pacientes com evidencia de invasão microscópica, a taxa de recorrência em 1,2 ano e 5 anos é de 20% e 33%, respectivamente, contrastando com nenhuma recorrência quando não se observava invasão microscópica (3). O tratamento de escolha é a remoção cirúrgica (taxa de mortalidade de 2 a 3% em mãos experientes), sendo necessária a tomas de medidas pré, intra e pós operatórias. No pré-operatório uso de bloqueadores alfa-adrenérgicos. Durante o intra-operatório cuidado na manipulação do tumor. No pós-operatório monitorar pressão arterial e níveis glicêmicos, pois pode ocorrer hipotensão e hipoglicemia (3). Nos casos de neoplasia maligna o tratamento clínico a longo prazo é necessário, já que ocorre impedimento de cirurgia. Este é realizado com o uso de bloqueadoresadrenergicos, e caso esses não sejam efetivos pode-se recorrer ao uso de metirosina(inibidor da tirosina-hidroxilase) que diminui a produção de catecolaminas pelo tumor. Embora os tumores malignos seja resistentes a radioterapia, a poliquimioterapia tem sido utilizada com limitado êxito (4). BIBLIOGRAFIA:
* Monitor e aluno do curso médico da Faculdade de Medicina da Bahia, Universidade Federal da Bahia, cursando o 5º semestre. |
_________________________________________________________________________________ |