| página inicial > monitoria > glicogenose |
| MONITORIA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discussão Anatomo-Clínica GLICOGENOSE |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Laudenor P. Lemos-Júnior* Q.P.: Aumento de volume abdominal há cerca de 5 anos. H.M.A: Mãe informa percepção de aumento no volume abdominal há cerca de 5 anos durante o curso de febre culminada em episódio convulsivo. A este tempo, foi realizada USG abdominal,sugerindo hepatomegalia inicial. Seguiu por pouco menos de três anos com aumento paulatino do volume abdominal associado a colúria, hipocolia fecal e episódios esporádicos de epistaxe. Foi encaminhado para o ambulatório de hepatologia (FAMEB) a partir de Barra Grande – BA, seguindo acompanhamento por 1 ano, com os mesmos sintomas, acrescido história de eventos hipoglicêmicos alternantes com eventos hiperglicêmicos (sem mais informações). Solicitada biópsia hepática há 2 anos, sugerindo glicogenose (excluido tipo IV ?). Mãe suspendeu tratamento há um ano, retornando no presente momento ao serviço por piora do quadro geral. Seguiu à internação por 2 dias para a realização de USG Abdominal, TC de Abdome, Laparotomia Exploradora e Biópsia Hepática e onde fez uso de medicação sintomática. I.S: Negou febre e icterícia. ANT. FISIOLÓGICOS: Pré-natal completo com todas as consultas, sem complicações. PNSV sem intercorrências, pesando 4400g. ANT. PATOLÓGICOS: Varicela resolvida há 3 anos. ANT. FAMILIARES: Hipercolesterolemia; HAS em avó materna. COBERTURA VACINAL: Cartão vacinal completo. CONDIÇÕES SÓCIO-ECONÔMICAS: Dificuldade financeira para vir até Salvador HABITAÇÃO: Casa rebocada; nunca viu barbeiro. HÁBITOS DE VIDA: Banhos freqüentes em rios e locais de água parada. EXAME FÍSICO Á INTERNAÇÃO
RESULTADOS DE PROVAS DIAGNÓSTICAS À INTERNAÇÃO
SUSPEITAS DIAGNÓSTICAS
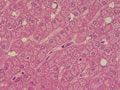
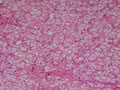
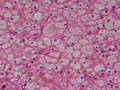
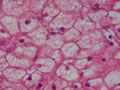
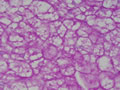
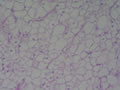
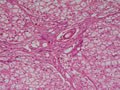
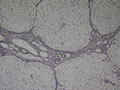
BIÓPSIA DE FÍGADO (06/10/2003):
RESUMO DA HISTÓRIA CLÍNICA: Trata-se de um paciente do sexo masculino, 6 anos, vítima de doença de armazenamento do glicogênio, cursando com quadro clássico de hepatomegalia, desde de ao menos 1 ano de idade. Há história de eventos hipo- e hiperglicêmicos alternantes e, porteriormente, hipocolia fecal, colúria e epistaxes. Foi detectado estado anêmico com neutropenia há última internação, juntamente com hipertrigliceridemia e transaminases elevadas. Negou febre e icterícia. O fígado é pálpavel a 4cm do RCD e indolor, sem esplenomegalia. USG e TC indicando hepatomegalia; biópsia hepática confirmando glicogenose. REVISÃO DA LITERATURA: As doenças de depósito do gligogênio (ou glicogenoses) são de cunho hereditário e causadas por anormalidade na síntese ou degradação do gligogênio (1). As concentrações de glicogênio >70mg/g, em tecido hepático, e / ou >15mg/g, em tecido muscular, caracterizam a glicogenose (2). Nos últimos tempos, os pacientes têm sido identificados pelos defeitos enzimáticos específicos, o que traz para nós hoje a caracterização de cada uma das formas da glicogenose, desde de que se identifique qual enzima está ausente ou insuficiente e se atente aos critérios clínicos particulares, como está na tabela 1 (2).
O glicogênio assume especial importância no estoque de glicose, sendo o responsável direto pela manutenção dos níveis estabilidados da glicemia de jejum, sendo o fígado o principal órgão de armazenamento, síntese e degradação (3). Pelo fato de dependermos da glicose vinda da dieta, é fácil presumir o quanto está susceptível a variações os pacientes com glicogenose (clique aqui para ver Figura 1) (1,2,3). Dentre os achados mais comuns estão, nestes grupos de pacientes, a hipoglicemia alternante com picos hiperglicêmicos, a hipertrigliceridemia, hiperlacticacidemia, transaminases elevadas e a hepatomegalia (1,3). Um aspecto relevante é que, dentre os problemas decorrentes do metabolismo indequado do glicogênio, encontramos a possibilidade de formação de moléculas de glicogênio anormais, as quais são resultado da deficiência em um dos três tipos enzimáticos: Enzimas Ramificadoras, Glicogênio Sintetase ou Enzimas Desramificadoras (2,3). A implicação prática está na maior associação da glicogenose com evolução para cirrose e / ou esplenomegalia (2). Dentre as complicações tadias destes pacientes estão:
O diagnóstico final é dado pela biópsia, ainda que a sintomatologia e a evolução sejam pilares fundamentais (1,2.3.4). A determinação dos componentes séricos envolvidos na patologia assume papel indispensável, embora a sua associação com a clínica não produza sensibilidade muito alta (2,4). O tratamento principal e fundamental consiste no controle dos níveis de glicemia com medidas não-farmacológicas (1,2). A intervenção cirúrgica (transplante) é indicada apenas para os pacientes com insuficiência hepática (1,2). BIBLIOGRAFIA:
*Monitor e aluno do curso médico da Faculdade de Medicina da Bahia, Universidade Federal da Bahia, cursando o 6º semestre. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
_________________________________________________________________________________ |